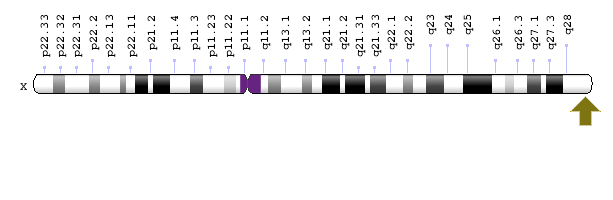
La duplication du gène MECP2 est causée par la duplication de l’ADN sur la région du chromosome Xq28 (28eme partie du bras long du chromosome X).
Ce n’est pas une maladie neurodégénérative inéluctable comme le syndrome de RETT.
Les problèmes d’occlusion sont dus à la duplication du gène voisin de MECP2 (FLNA – Filamine), un autre gène est également responsable de désordre : Irak1. Selon certaines études, ce gène serait responsable d’un déficit immunitaire et d’une grande fragilité pulmonaire.
La région Xq28 contient plusieurs gènes dont le MECP2 et IRAK1, GDI1, FLNA, L1CAM, IDH3G. Le début et la fin en double (points d’arrêt) varient entre les individus. La constatation est désormais établie que la duplication du gène MecP2 joue un rôle important dans les troubles du système nerveux. Des souris génétiquement modifiées avec 2 fois le niveau normal de la proteine MecP2, développent les caractéristiques du syndrome de duplication. Ces études démontrent bien que MecP2 (plutôt que d’autres proteines) est le coupable de cette anomalie. MecP2 a aussi un autre effet : Plus la proteïne est produite, moins les patients ressentent la douleur.
Certains enfants présentent également des anomalies (duplication) sur le chromosome Xq26.3 comprenant 23 gènes.
Dans plus de 90% des cas, la duplication est héritée d’une mère porteuse asymptomatique. On parle de porteur lorsque la personne est porteuse du défaut génétique, mais que celui-ci ne s’exprime pas cliniquement.
Lors d’une grossesse, la mère a 50% de risque de faire naître un garçon atteint et 50% de mettre au monde une fille porteuse de la mutation. Il est très rare que les filles développent une forme symptomatique de la maladie.
Les hommes (XY), ne possèdent qu’un seul chromosome X. La duplication du gène MECP2 cause dans tous les cas des symptômes graves.
Contrairement aux hommes, les femmes (XX) possèdent 2 chromosomes X. Une duplication du gène MECP2 a très rarement des conséquences sur une femme, car le chromosome X ayant du matériel dupliqué est inactivé et le taux de protéine MeCP2 reste normal.
Dans de rares cas, la mutation est exprimée chez les femmes à cause d’une inactivation défavorable du chromosome X muté, ou parce que la mutation est transposée sur un autre chromosome.
La duplication du gène MECP2 peut également apparaitre lors de la fécondation. On parle alors de mutation de novo car aucun des parents n’est porteur de la mutation génétique.
En terme d’espérance de vie, différentes études démontrent une mortalité importante vers la vingt-cinquième année. Ce n’est pas la duplication qui en est la cause mais les pathologies (infections respiratoires principalement) qui en sont responsables. Nous avons aussi rencontré des porteurs de l’anomalie de plus de 40 ans.


